
Enfermedad de Huntington y otras enfermedades del sistema nervioso central
Resumen de Investigación:
Estudiamos los mecanismos moleculares subyacentes a las enfermedades neurales generando modelos genéticos de ratón (como el modelo de Huntington HD94 Cell 101:57-66 2000 citado 972 veces, el modelo de Alzheimer Tet/GSK3 EMBO J 20:27-39 2001 citado 870 veces, o el Modelo TgCPEB4Δ4 de autismo idiopático Nature 560:441-446 2018 citado 89 veces) para validar vías patogénicas y probar nuevas estrategias terapéuticas.
La enfermedad de Huntington (EH) es una enfermedad neurodegenerativa autosómica dominante caracterizada por movimientos involuntarios, síntomas psiquiátricos y demencia. Es causada por una expansión del trinucleótido CAG ubicado en el gen de la huntingtina. Al caracterizar las interacciones moleculares del ARN y la proteína mutados, y en particular su impacto en las proteínas de unión al ARN y el procesamiento del ARN, encontramos nuevas pistas sobre la EH y otros trastornos neurales como la distonía con parkinsonismo ligada al cromosoma X (XDP), la epilepsia, el autismo y la esquizofrenia.
Por ejemplo, al caracterizar la alteración global del splicing en el estriado de la EH (Brain 143: 2207-19, 2020 y Brain 144:2009-23, 2021), encontramos una alteración de tau (la proteína que se acumula dentro de las neuronas en la enfermedad de Alzheimer) . Mediante el análisis de modelos de ratones transgénicos con EH con niveles variables de tau, demostramos que tau contribuye a la patogénesis de la EH y descubrimos un nuevo sello histopatológico (los bastones nucleares de Tau o hendiduras nucleares de Tau) (Nat Med. 2014, 20:881-5 citado 195 veces).
Al caracterizar el papel de la poliadenilación citoplasmática de los ARNm por parte de las CPEBs en la EH, descubrimos una deficiencia tratable de tiamina en los cerebros de EH (Sci Transl Med. 2021 13:eabe7104) que ha originado un ensayo clínico (https://clinicaltrials.gov/ct2/show /NCT04478734).
También encontramos que la mayoría de los genes de riesgo del trastorno del espectro autista (TEA) son diana de CPEB4. De hecho, CPEB4 muestra un desequilibrio de isoformas en personas con TEA debido al splicing incorrecto de un microexón específico neuronal. En consecuencia, los ratones con un desequilibrio equivalente de CPEB4 muestran un fenotipo similar al autismo (Nature 560:441-446, 2018) y se han convertido en un modelo útil de autismo idiopático.
Recientemente, también hemos descrito una alteración de la poliadenilación citoplasmática en la epilepsia (Brain 143: 2139-53 2020).
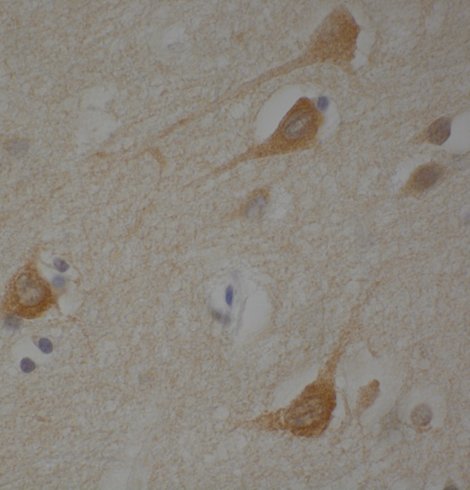
Tau Nuclear Rods (TNRs) en neuronas de un paciente de EH.
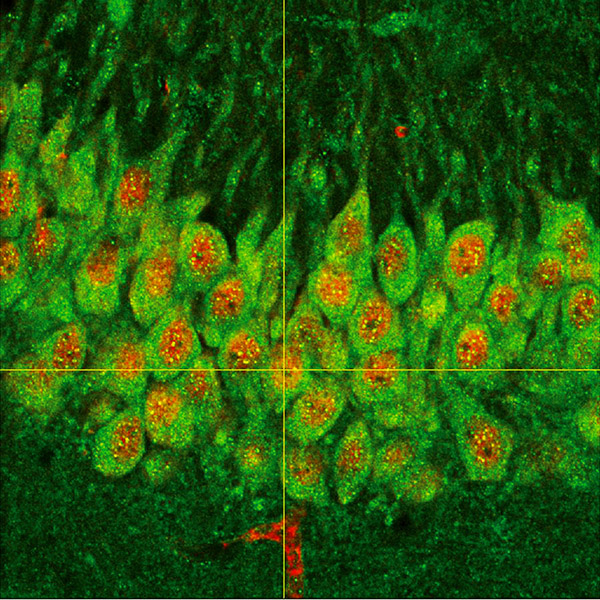
Expresión del factor de transcripción ATF5 en neuronas de hipocampo de ratón.

| Apellidos | Nombre | Laboratorio | Ext.* | Categoría profesional | |
|---|---|---|---|---|---|
| Arroyo García | Alejandra María | 209 | 4582 | amarroyo(at)cbm.csic.es | M3 Predoc.formación |
| Coiduras del Olmo | Marta | 209 | 4552 | Becario JAE Intro | |
| González Bermejo | María | 209 | 4452 | M3 Predoc.formación | |
| Khachatryan Boyajyan | Avetik Arsen | 209 | 4582 | Estudiante TFG | |
| Lozano Muñoz | David | 209 | 4552 | dlozano(at)cbm.csic.es | Titulado Sup. Actividades Tecn. y Prof.GP1 |
| Lucas Lozano | José Javier | 209 | 4552 | jjlucas(at)cbm.csic.es | E. Profesores de Investigación de Organismos Públicos de Investigación |
| Lucas Santamaría | Miriam | 209 | 4582 | mmlucas(at)cbm.csic.es | Contratado CIBER |
| Pose Utrilla | Julia | 209 | 4582 | jpose(at)cbm.csic.es | Titulado Superior Grado de Doctor |
| Rodríguez López | Claudia | 209 | 4582 | crodriguez(at)cbm.csic.es | M3 |
| Ruiz Blas | Irene | 209 | 4582 | M2 |
Publicaciones relevantes:
- Ollà I, Pardiñas AF, Parras A, Hernández IH, Santos-Galindo M, Picó S, Callado LF, Elorza A, Rodríguez-López C, Fernández-Miranda G, Belloc E, Walters JTR, O'Donovan MC, Méndez R, Toma C, Meana JJ, Owen MJ, Lucas JJ (2023) Pathogenic mis-splicing of CPEB4 in schizophrenia. Biol Psychiatry. DOI: 10.1016/j.biopsych.2023.03.010.
- Picó S, Parras A, Santos-Galindo M, Pose-Utrilla J, Castro M, Fraga E, Hernández IH, Elorza A, Anta H, Wang N, Martí-Sánchez L, Belloc E, Garcia-Esparcia P, Garrido JJ, Ferrer I, Macías-García D, Mir P, Artuch R, Pérez B, Hernández F, Pérez-Cerdá C, Navarro P, López-Sendón JL, Iglesias T, Yang XW, Méndez R, Lucas JJ (2021) CPEB alteration and aberrant transcriptome-polyadenylation lead to a treatable SLC19A3 deficiency in Huntington’s disease. Sci. Transl. Med. DOI: 10.1126/scitranslmed.abe7104
- Elorza A, Marquez Y, Cabrera JR, Sanchez-Trincado JL, Santos-Galindo M, Hernandez IH, Diaz-Hernandez JI, Garcia-Escudero R, Irimia M and Lucas JJ (2021) Huntington’s disease-specific mis-splicing unveils key effector genes and altered splicing factors. Brain 144:2009-23
- Hernández IH, Cabrera JR, Santos-Galindo M, Sánchez-Martín M, Domínguez V, García-Escudero R, Pérez-Álvarez MJ, Pintado B and Lucas JJ (2020) Pathogenic SREK1 decrease in Huntington’s disease lowers TAF1 mimicking X-linked dystonia parkinsonism. Brain 143:2207-19
- Parras A, de Diego-Garcia L, Alves M, Beamer E, Conte G, Jimenez-Mateos EM, Morgan J, Ollà I, Hernandez-Santana Y, Delanty N, Farrell MA, O’Brien DF, Ocampo A, Henshall DC, Méndez R, Lucas JJ* and Engel T* (2020) mRNA polyadenylation as a novel regulatory mechanism of gene expression in temporal lobe epilepsy. Brain 143:2139-53
- Parras A, Anta H, Santos-Galindo M, Swarup V, Elorza A, Nieto-González JL, Picó S, Hernández IH, Díaz-Hernández JI, Belloc E, Rodolosse A, Parikshak NN, Peñagarikano O, Fernández-Chacón R, Irimia M, Navarro P, Geschwind DH, Méndez R, Lucas JJ. (2018) Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing. Nature 560:441-6
- Hernández IH, Torres-Peraza J, Santos-Galindo M, Ramos-Morón E, Fernández-Fernández MR, Pérez-Álvarez MJ, Miranda-Vizuete A, Lucas JJ. (2017) The neuroprotective transcription factor ATF5 is decreased and sequestered into polyglutamine inclusions in Huntington's disease. Acta Neuropathol. 134:839-50
- McKinnon C, Goold R, Andre R, Devoy A, Ortega Z, Moonga J, Linehan JM, Brandner S, Lucas JJ, Collinge J and Tabrizi SJ. (2015) Prion-mediated neurodegeneration associated with early impairment ofubiquitin-proteasome system. Acta Neuropathol. 131:411-25
- Fernández-Nogales M, Cabrera JR, Santos-Galindo M, Hoozemans JJM, Ferrer I, Rozemuller AJM, Hernández F, Avila J and Lucas JJ (2014) Huntington’s disease is a four-repeat tauopathy with tau-nuclear rods. Nat Med 20, 881-5
- Torres-Peraza JF, Engel T, Martín-Ibañez R, Fernández-Fernández MR, Esgleas M, Canals JM, Henshall DC & Lucas JJ (2013) Protective neuronal induction of ATF5 in endoplasmic reticulum stress induced by status epilepticus. Brain 136:1161-76
- Gomez-Sintes R and Lucas JJ (2010) NFAT/Fas-signaling mediates apoptosis and neurological side effects of GSK-3 inhibition in a mouse model of lithium therapy. J Clin Invest 120:2432-45
- Maynard CJ, Böttcher C, Ortega Z, Smith R, Florea BI, Diaz-Hernandez M, Brundin P, Overkleeft H, Li JY, Lucas JJ & Dantuma N (2009) Accumulation of ubiquitin conjugates in a polyglutamine disease model occurs without global ubiquitin/proteasome system impairment PNAS 106:13986-91
- Gómez-Sintes R, Hernández F, Bortolozzi A, Artigas F, Avila J, Zaratin P, Gotteland J-P & Lucas JJ (2007) Neuronal apoptosis and reversible motor deficit in dominat-negative GSK-3 conditional transgenic mice. EMBO J. 26: 2743-54
- Lucas JJ, Hernández F, Gómez-Ramos P, Morán MA, Hen R, and Avila J (2001) Decreased nuclear ß-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3ß conditional transgenic mice. EMBO J. 20, 27-39
- Yamamoto A*, Lucas JJ* & Hen R (2000) *These authors contributed equally to the work. Reversal of neuropathology and motor dysfunction in a conditional model of Huntingtron´s Disease. Cell 101: 57-66
Otras actividades:
- Grupo integrante del Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED)
- José Lucas nombrado Académico Correspondiente de la Real Academia Nacional de Farmacia, Junio de 2011


















